Publications
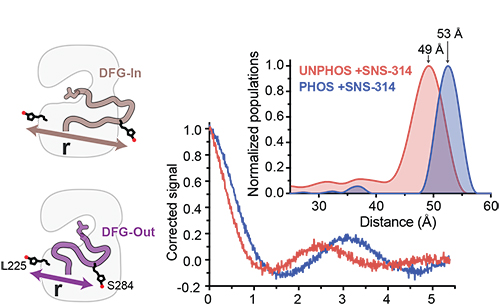
Allostery governs Cdk2 activation and differential recognition of CDK inhibitors
Majumdar, A., Burban, D.J., Muretta, J.M., Thompson, A.R., Engel, T.A., Rasmussen, D.M., Subrahmanian, M.V., Veglia, G., Thomas, D.D., Levinson, N.M. Nature Chemical Biology 2021.
We used a combination of double electron-electron resonance (DEER) spectroscopy, paramagnetic relaxation enhancement NMR, and fluorescence to study allosteric activation of cyclin-dependent kinase 2 in solution. We showed that phosphorylation of Cdk2 on the activation loop enhances allosteric coupling between the cyclin subunit and the kinase, and demonstrate that this allosteric coupling underlies the selective recognition of CDK inhibitors.
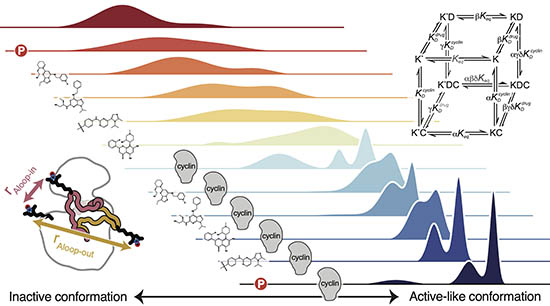
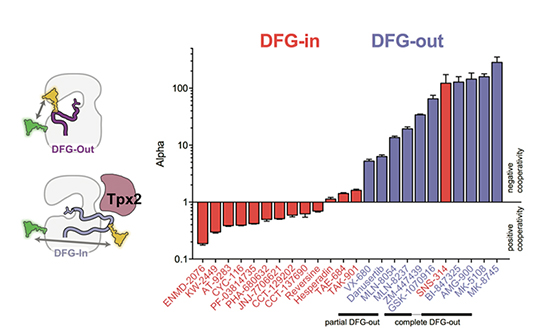
Quantitative conformational profiling of kinase inhibitors reveals origins of selectivity for Aurora kinase activation states
Lake, E.W., Muretta, J.M., Thompson, A.R., Rasmussen, D.M., Majumdar, A., Faber, E.B., Ruff, E.F., Thomas, D.D., Levinson, N.M. PNAS 2018.
We used a new time-resolved FRET based method to profile the conformational effects of a large panel of Aurora kinase inhibitors on Aurora A. The results show clearly for the first time that the preference of inhibitors for the DFG-in or DFG-out states is the dominant determinant of selectivity, and that different DFG-out inhibitors differ by two orders of magnitude in their preference for the DFG-out state. This suggests that many of these inhibitors selectively target different cellular pools of Aurora A, with important implications for the clinical use of Aurora A inhibitors currently in trials.
A dynamic mechanism for allosteric activation of Aurora kinase A by activation loop phosphorylation
Ruff, E.F., Muretta, J.M., Thompson, A.R., Lake, E.W., Cyphers, S., Albanese, S.K., Hanson, S.M., Behr, J.M., Thomas, D.D., Chodera, J.D., Levinson, N.M. eLife 2018.
We used a diverse range of spectroscopic methods combined with long timescale molecular dynamics simulations to show that phosphorylation of the activation loop does not trap Aurora kinase A in the active DFG-in state as previously thought, and that the loop instead continues to transition between the DFG-in and DFG-out states. Activation occurs through tuning of the catalytic activity of the DFG-in subpopulation through a phosphorylation-driven switch from a previously undetected autoinhibited DFG-in substate to the active DFG-in substate.
A water-mediated allosteric network governs activation of Aurora kinase A
Cyphers, S., Ruff, E., Behr, J., Chodera, D.J., Levinson, N.M. Nature Chemical Biology 2017.
We used an infrared probe to show that allosteric activation of the mitotic kinase Aurora A by the spindle-associated protein Tpx2 is a two-step process, involving both a conformatonal shift towards the active DFG-In state, and an enhancement of the intrinsic activity of the DFG-In state through tuning of a novel allosteric network mediated by structured water molecules.

A conserved water-mediated hydrogen bond network defines bosutinib's kinase selectivity
Levinson, N.M. and Boxer, S.G. Nature Chemical Biology 2014. 10,127-132.
Type I kinase inhibitors exploit structured water molecules in the ATP-binding site to recognize their kinase targets. These interactions turn out to be energetically significant and make a big contribution to the selectivity that inhibitors display for different kinases.
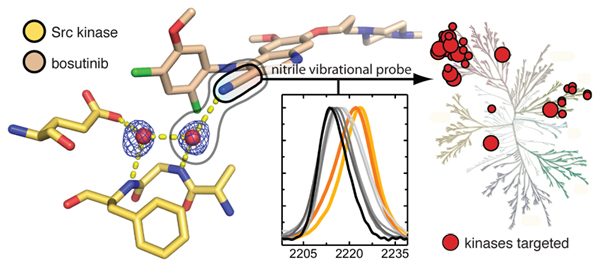
Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain
Levinson, N.M. and Boxer, S.G. PLoS ONE 2012. 10,127-132.
Bosutinib is a second-generation Abl kinase inhibitor used in the treatment of chronic myeloid leukemia. We determined the first crystal structure of this drug bound to the Abl kinase domain. The structure explains the effects of several resistance mutations that arise in patients during tyrosine kinase inhibitor therapy.

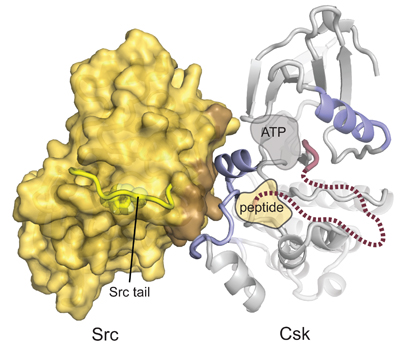
Structural basis for the recognition of c-Src by its inactivator, Csk
Levinson, N.M., Seeliger, M., Cole, P.A., Kuriyan, J. Cell 2008. 10,127-132.
Src kinase, the prototypical oncogene, is catalytically inactivated through phosphorylation on a C-terminal tyrosine residue. The kinase responsible, Csk, only phosphorylates the Src kinases. We illuminated the structural basis for this unusual specificity by cocrystallizing the two proteins.
A Src-like inactive conformation in the Abl tyrosine kinase domain
Levinson, N.M., Kuchment, O., Shen, K., Young, M.A., Koldobskiy, M., Karplus, M., Cole, P.A., Kuriyan, J. PLoS Biology 2006. 4,144.
The distantly related tyrosine kinases Src and Abl were previously thought to have unrelated allosteric activation mechanisms. Here we showed that Abl can in fact adopt an inactive conformation that looks almost identical to the canonical inactive conformation of Src, demonstrating that the conformational states that protein kinases adopt are broadly conserved across this protein family.
© Levinson Lab 2014 | University of Minnesota, Twin Cities | Department of Pharmacology | 312 Church Street SE, Minneapolis, MN 55455